



Vijay Kumarover 2 years ago
Cystic Fibrosis
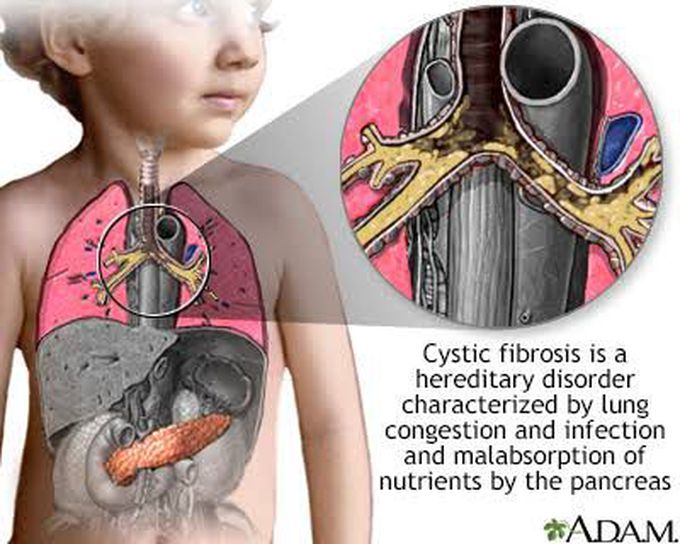
Cystic Fibrosis is an inherited autosomal recessive disease of adults and children. In which there is mutation of CFTR gene on chromosome 7 which results in thick viscous secretion, impaired mucus clearance in the lung, impaired pancreatic secretion and abnormal sweat. Clinical features include respiratory symptoms ( productive cough, chronic bronchitis and bronchiectasis, digital clubbing) , intestinal symptoms (meconium ileus), pancreatic insufficiency ( DM, fat soluble vitamins deficiency, steatorrhea). Diagnostic test include sweat test, CXR, DNA testing and nasal potential difference. Management promotes clearance of airway secretion, antibiotics, pancreatic enzymes replacement and fat soluble vitamins replacement.
Other commentsSign in to post comments. You don't have an account? Sign up now!
Related posts
Hereditary Mucoepithelial DysplasiaHereditary Hemorrhagic Telangiectasia (HHT).Hereditary Hemorrhagic Telangiectasia (HHT).Types of Congenital Heart DefectsDiagrammatic representation of pathogenesis of hereditary spherocytosisHereditary spherocytosis.Cystic Fibrosis IHereditary causes of unconjugated hyperbilirubinemiaHyperekplexiaFabry disease
