



Creutzfeldt–Jakob Disease
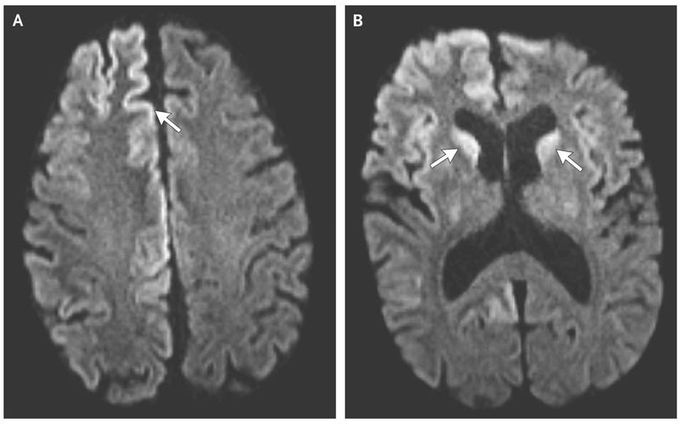
A 54-year-old man presented to the emergency department with a 3-week history of cognitive deterioration and functional decline. He was disoriented, inattentive, and disinhibited. Neurologic examination revealed horizontal gaze-evoked nystagmus, hyperreflexia on the left side, startle myoclonus, and ataxia. Magnetic resonance imaging (MRI) revealed hyperintensity of the cortical gyri of the frontal and occipital lobes, called cortical ribboning (Panel A, arrow), and within the caudate heads on both sides (Panel B, arrows) on diffusion-weighted imaging. Electroencephalography revealed lateralized periodic discharges in the right hemisphere without evidence of seizures. Testing of cerebrospinal fluid was positive for 14-3-3 and tau proteins, which are known markers of prion disease, and the real-time quaking-induced conversion assay, which is highly sensitive for the detection of the abnormal form of the prion protein, was also positive. Given the patient’s rapid clinical deterioration, imaging findings, and cerebrospinal fluid markers, he received a diagnosis of Creutzfeldt–Jakob disease, a fatal spongiform encephalopathy that is caused by the accumulation of abnormal prions. Creutzfeldt–Jakob disease leads to cognitive decline and is characterized by additional neurologic features that manifest according to the location of lesions. Common MRI findings include hyperintensity of the cortex, pulvinar and dorsomedial thalamic nuclei, and basal ganglia on T2-weighted, diffusion-weighted, and fluid-attenuated inversion recovery sequences. The patient died in hospice 8 weeks after the initial onset of symptoms.
Absolutely the scariest case I was ever involved in was a biopsy to confirm CJD.......nowadays there are disposable kits to do a burrhole biopsy but the thought that everything even the metal instruments had to be incinerated was very disconcerting and scary


