



Gaucher’s Disease
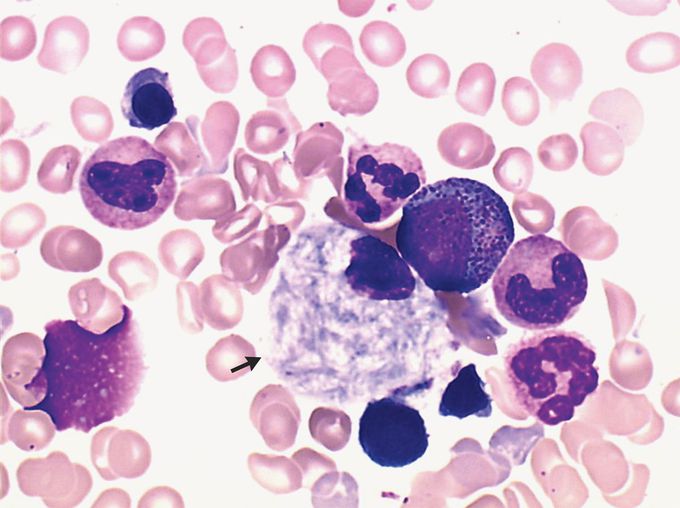
A 73-year-old man presented to the emergency department with a 1-month history of fatigue and diffuse bone pain. He reported no abdominal pain or neurologic symptoms. On physical examination, there was no hepatosplenomegaly. Laboratory studies showed a white-cell count of 1700 per cubic millimeter (reference range, 4000 to 10,000), a hemoglobin level of 5.9 g per deciliter (reference range, 12 to 18), and a platelet count of 111,000 per cubic millimeter (reference range, 120,000 to 180,000). Whole-body magnetic resonance imaging revealed osteonecrosis of the right humerus and right femur. Bone marrow biopsy revealed macrophages with a “wrinkled tissue paper” appearance in the cytoplasm (arrow, May–Grünwald Giemsa staining), a finding consistent with Gaucher’s cells. Further testing identified reduced β-glucocerebrosidase activity and homozygosity with the N370S variant of the GBA gene. Workup for hematologic cancers was negative. A diagnosis of type 1 Gaucher’s disease was made. Gaucher’s disease is an autosomal recessive disease that results in a deficiency of the lysosomal enzyme β-glucocerebrosidase. In type 1 Gaucher’s disease, some patients do not present with symptoms until late adulthood. Clinical features include fatigue, hepatosplenomegaly, pancytopenia, bone pain, osteonecrosis, and — in children — growth retardation. The patient began receiving enzyme-replacement therapy. At 6 months of follow-up, his symptoms had abated and cell counts had improved.
