



PEELING SKIN SYNDROME (PSS)
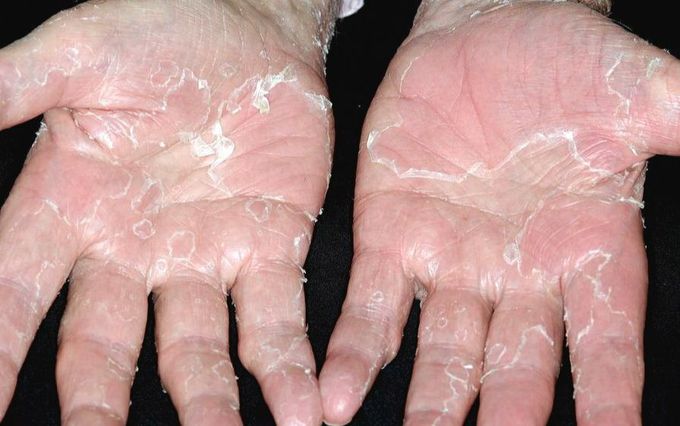
Peeling skin syndrome (PSS) is a group of rare inherited skin disorders in which the normal gradual process of invisible shedding of the outermost skin layers is hastened and/or aggravated. PSS is characterized by painless, continual, spontaneous skin peeling (exfoliation) due to a separation of the outermost layer of the epidermis (stratum corneum) from the underlying layers. Other findings may include blistering and/or reddening of the skin (erythema) and itching (pruritus). Symptoms may be present from birth or appear in early childhood and are often exacerbated by friction, heat or other external factors. Based on the extent of skin involvement, PSS may involve the skin of the entire body (generalized form), or is limited to the extremities, mostly hands and feet (localized form). Generalized PSS can be distinguished into an inflammatory type which is associated with erythema, involves other organ systems and is more severe, and a milder, non-inflammatory type. PSS may be caused by disease-causing variants in multiple genes encoding proteins with crucial functions for cell-cell adhesion: structural proteins forming cell-cell adhesion points (desmosomes, corneodesmosomes) and inhibitors of epidermal proteases that control skin shedding. Signs & Symptoms Peeling skin syndrome belongs to the groups of congenital ichthyosis and skin fragility disorders with autosomal recessive inheritance. Most forms of PSS manifest at birth or during infancy with shedding or peeling of the outermost layer of the skin (horny layer, aka stratum corneum). Skin peeling occurs spontaneous, is painless, and may persist lifelong with gradual improvements. Often, affected individuals and/or their caregivers can remove sheets of skin manually, comparable to skin peeling after a severe sunburn. Other findings associated with this disorder may include blistering and skin fragility, itching, short stature, and/or newly formed hairs that can be plucked out more easily than normal. Skin peeling is often exacerbated by mechanical irritation of the skin, heat, sweat or water exposure or other external factors. In the localized types, individuals develop blisters and erosions on hands and feet at birth or during infancy, which is reminiscent of another blistering skin disorder, epidermolysis bullosa simplex. The generalized inflammatory types, such as SAM syndrome or Netherton syndrome may be associated with generalized inflammation of the skin (erythroderma) or localized thickened, red plaques (erythrokeratoderma), immunodysfunction with elevated IgE levels, allergies, and susceptibility to infections, failure to thrive or metabolic wasting. In some patients, these disorders may be life-threatening, especially during the newborn period. Due to the variable clinical presentations of PSS, its often mild features and gradual improvement with age, PSS may be underdiagnosed and underreported. Synonyms of Peeling Skin Syndrome deciduous skin familial continuous skin peeling skin peeling syndrome exfoliative ichthyosis PSS Subdivisions of Peeling Skin Syndrome generalized non-inflammatory type (PSS6, PSS5, PSS3) generalized inflammatory type (PSS1) localized (acral) type (PSS2, PSS4) Causes To date, genetic changes in several distinct genes have been reported to cause PSS. These genes encode either structural proteins of corneocytes, the cells of the outermost skin layer (CDSN; DSG1; FLG2; DSC3; JUP) or inhibitors of epidermal proteases (SPINK5, CSTA; CAST; SERINB8), which are crucial regulators for the degradation of corneodesmosomes and shedding of corneocytes. Generalized non-inflammatory type FLG2: The filaggrin 2 gene (FLG2) is co-expressed with corneodesmosin (CDSN, see below) in the outermost layers of the skin, where it is cleaved into multiple small repeat units and is crucial for maintaining cell-cell adhesion. Complete or almost complete filaggrin 2 deficiency due to loss-of-function variants in FLG2 results in decreased expression of CDSN, and generalized, non-inflammatory PSS. The generalized dryness and peeling of the skin typically improves with age but can be triggered or aggravated by heat exposure, mechanical trauma to the skin and other external factors. Rarely, formation of blisters has been reported. CAST: This gene encodes calpastatin, an endogenous protease inhibitor of calpain, which plays a role in various cell functions such as cell proliferation, differentiation, mobility, cell cycle progression, and apoptosis. Several homozygous loss-of-function variants in the CAST gene have been reported in association with PLACK syndrome, an autosomal recessive form of generalized peeling skin syndrome associated with leukonychia (white nails), acral punctate keratoses and knuckle pads (small, callus-like plaques of thickened skin on palms and soles and over knuckles), and angular cheilitis (inflammation on the corners of the mouth). Skin peeling manifests in infancy and improves over time, although it may worsen with heat exposure in the summer. The features may overlap with pachyonychia congenita, including oral leukokeratosis (whitish thickened plaques inside the mouth), and more diffuse plantar keratoderma. SERPINB8: The SERPINB8 gene codes for an epidermal serine protease inhibitor, which is, similar to SPINK5 involved in Netherton syndrome, crucial for balance between cell-cell adhesion and shedding of corneocytes. Different homozygous variants in the SERPINB8 gene have been reported in three unrelated families with autosomal recessive peeling skin syndrome, with evidence of reduced protein expression and altered cell adhesion in affected skin. The affected individuals presented in infancy with peeling of the skin of varying severity, with or without erythema or hyperkeratotic plaques on the palms and soles. CHST8: Function of the carbohydrate sulfotransferase gene CHST8 and its role in human disease have not been completely established. A homozygous missense variant in the CHST8 gene has been reported in multiple individuals with generalized non-inflammatory peeling skin syndrome from a single large consanguineous family. While initial studies suggested that the reported variant results in decreased expression and loss of function, these findings were not confirmed by functional follow-up studies, suggesting another, not yet identified, genetic cause of PSS in that family. Generalized inflammatory type CDSN: PSS1 is caused by deleterious loss-of-function changes in the corneodesmosin gene, CDSN. Corneodesmosin is a secreted glycoprotein and main component of cell-cell adhesion structures called corneodesmosomes within the stratum corneum and is also found in hair follicles. While complete loss of corneodesmosin due to variants on both alleles of the gene (bi-allelic) results in generalized peeling skin syndrome, abnormal corneodesmosin (due to sequence changes on only one copy of the gene) have been described in the autosomal dominant hair disorder ‘hypotrichosis simplex (MIM146520)’. In Japanese individuals, a particularly common cause of PSS1 is a genomic deletion of 6 genes including the entire CDSN gene. DSG1: DSG1 codes for desmoglein 1, a major structural protein of desmosomal plaques and corneodesmosomes forming adhesion junctions between cells. Complete loss of desmoglein 1 due to bi-allelic pathogenic variants in DSG1 typically causes SAM syndrome, characterized by severe skin dermatitis, multiple allergies and metabolic wasting, which can be life-threatening. Nevertheless, there is a broad range of features and severity, and some DSG1 variants, especially those in the repeat unit domain, may result in a milder phenotype with features of PSS. SPINK5: Features of generalized inflammatory peeling skin syndrome may be present in individuals with Netherton syndrome, which is caused by bi-allelic loss-of-function variants in the SPINK5 gene. NTS is characterized by a triad of erythema and peeling of the skin associated with characteristic hair shaft abnormalities (“bamboo hair’), and immunodysregulation with elevated IgE levels, atopy and allergies. SPINK5 codes for the multidomain protease inhibitor LEKTI, crucial for checking proteolytical activity of several epidermal serine proteases. Its loss leads to increased degradation of corneodesmosomes and excessive shedding of corneocytes, abnormal formation of the lipid envelope, and to a severe skin barrier dysfunction aggravated by a loss of crucial anti-inflammatory and anti-microbial protection normally provided by LEKTI. Localized type TGM5: The transglutaminase-5 (TGM5) gene is responsible for the acral, localized form of peeling skin syndrome. Bi-allelic disease-causing genetic variants in TGM5 result in loss of function of this important cross-linking enzyme in the upper skin layers. Acral PSS presents with superficial blistering and peeling on the inner and outer surfaces of hands and feet, sometimes resembling the blistering skin disorder epidermolysis bullosa simplex. The majority of affected individuals reported to date harbor the common missense variant p.Gly113Cys in the TGM1 gene, either in a heterozygous or homozygous state. This recurrent variant is especially common and disseminated across the European continent, and has also been observed homozygously in the general population, suggesting that acral PSS is more common and underdiagnosed due to its mild features. CSTA: The cystatin A gene encodes a cysteine proteinase inhibitor expressed in the outermost skin layers. This enzyme is, similar to other protease inhibitors, an important player in cell-cell adhesion. Biallelic loss-of-function variants in CSTA may also cause acral peeling skin syndrome. Major features are dry, scaly skin with hyperhidrosis, erythroderma, and peeling on palms and soles aggravated by heat, friction, and water or sweat exposure. All known forms of peeling skin syndrome are inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits two abnormal copies of the disease gene, usually one from each parent. If an individual receives one normal gene copy and one abnormal gene copy for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene copy and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females. All individuals are thought to be carriers for at least 4-5 abnormal recessive genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder. Affected Populations Peeling skin syndrome is a rare inherited disorder that, in theory, affects males and females in equal numbers. More than 100 cases have been reported in the medical literature from different population groups. Peeling skin syndrome due to variants in the CHST8 and CSTA genes were reported in consanguineous Pakistani and in Bedouin families, respectively. In Japanese individuals, a particularly common deletion involving CDSN has been reported. In Europeans, acral PSS due to TMG5 variants is more common. Diagnosis A good history and physical exam are often sufficient to make the diagnosis, although specialized tests including surgical removal and microscopic evaluation (biopsy) of affected tissue may be necessary at times. The continual shedding of large sheets of skin distinguishes peeling skin syndrome from Netherton syndrome and from other types of autosomal recessive congenital ichthyosis, such as congenital ichthyosiform erythroderma. The skin of so-called “collodion babies” peels off after a few weeks and does not return, in contrast to patients with peeling skin syndrome whose symptoms return time after time. Standard Therapies Treatment Treating peeling skin syndrome by applying skin softening (emollient) ointments, especially after a bath while the skin is moist, may offer some relief. Plain petroleum jelly or Vaseline is preferred. None of the corticosteroids or systemic retinoids (vitamin A derivatives) is indicated or effective and all may have serious side effects or adverse reactions. To discuss the risk of having children with this disorder and the possibility of genetic testing, genetic counseling is recommended for affected individuals and their families.

Thank you so much ,for the usefully information, i have this condition now i know how to deal with it
