



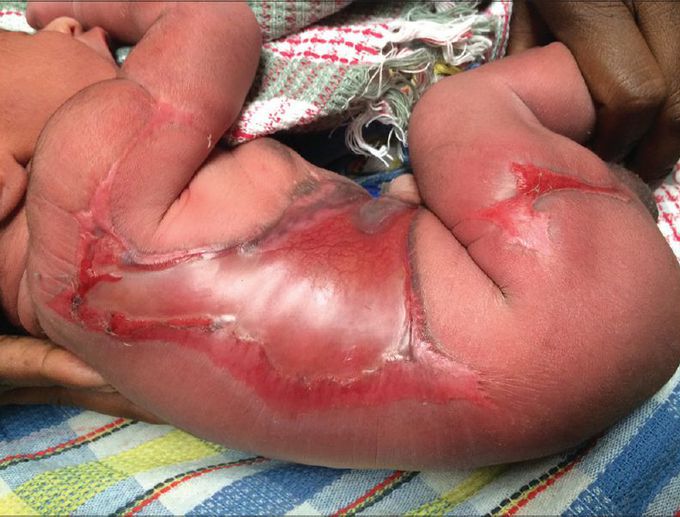
APLASIA CUTIS CONGENITA (ACC)
Aplasia Cutis Congenita is a rare disorder with a complicated pattern of inheritance. Babies are born with the absence of certain layer(s) of skin, most often on the scalp, but also on the trunk, and/or arms and legs. The affected area is typically covered with a thin, transparent membrane. The skull and/or underlying areas may be visible and be abnormally developed. Aplasia Cutis Congenita may be the primary disorder or it may occur in association with other underlying disorders. Medical treatments of Aplasia Cutis Congenita include measures to prevent the drying out of the membrane by soothing, bland ointments. Antibiotics should be used only if signs of bacterial infection are present. The damaged area usually heals spontaneously. classification system of aplasia cutis congenita consisting of 9 groups based on the location of the lesions and the presence or absence of associated malformations: Group 1 This is scalp aplasia cutis congenita without multiple anomalies. Nearly 86% of all solitary lesions occur on the scalp. A collar of hair is often seen around the defect, particularly with membranous aplasia cutis. It can be autosomal dominant or sporadic. Group 2 This is scalp aplasia cutis congenita with limb anomalies. Adams-Oliver syndrome is a distinct disorder in which distal limb reduction abnormalities are found in association with solitary midline scalp defects. Adams-Oliver syndrome exhibits both autosomal dominant and autosomal recessive patterns of inheritance. In recent years, mutations in EOGT and DOCK6 have been identified as causes for autosomal recessive Adams-Oliver syndrome, and mutations in DLL4,ARHGAP31, RBPJ, and NOTCH1 have been observed in autosomal dominant forms of the syndrome. In this group of aplasia cutis congenita, the scalp lesions tend to be large. The most common limb malformation is hypoplastic or absent distal phalanges, but the severity of limb anomalies ranges from minor defects such as an absent nail or broad fingertip to more severe involvement. Limb anomalies are usually asymmetric and more commonly involve the lower extremities. Other anomalies may include cutis marmorata telangiectatica congenita, hemangiomas, cranial arteriovenous malformation, congenital heart defects, skin tags, supernumerary nipples, and woolly hair. Group 3 This is scalp aplasia cutis congenita with epidermal and sebaceous (organoid) nevi,also involving the scalp usually adjacent to the aplasia cutis. Some patients have also had ophthalmic and neurologic findings typical of epidermal nevus syndrome, including seizures, mental retardation, corneal opacities, and eyelid colobomas. SCALP syndrome has been proposed as an entity to describe the constellation of nevus Sebaceus, Central nervous system malformations, Aplasia cutis congenita, Limbal dermoid, and Pigmented nevus. Inheritance is sporadic. Group 4 This is aplasia cutis congenita overlying deeper embryologic malformations.Examples include meningomyelocele, porencephaly, leptomeningeal angiomatosis, cranial stenosis, spinal dysraphism, gastroschisis, and omphalocele. A hair collar is often present in the scalp lesions overlying neural tube defects. Intracranial arteriovenous malformations and fistulas have also been reported in association with scalp aplasia cutis congenita in rare cases.The inheritance pattern in this group varies with the associated underlying condition. There is often a need for abdominal wall defect repair with this type of aplasia cutis congenita. Group 5 This is aplasia cutis congenita associated with fetus papyraceous or placental infarct.Fetus papyraceous is found at the time of delivery and results from the death of a twin fetus in the late first or early second trimester. The surviving fetus is affected with extensive truncal and limb aplasia cutis congenita in a linear or stellate configuration but is usually otherwise normal. Group 6 This is aplasia cutis congenita associated with epidermolysis bullosa (EB), also referred to as Bart syndrome. Aplasia cutis congenita can be seen with any type of EB (simplex, junctional, or dystrophic). Many reports describe aplasia cutis congenita usually occurring on the lower extremities. A subgroup includes the association of pyloric or duodenal atresia, ureteral stenosis, renal abnormalities, craniofacial abnormalities, nail dystrophy, and aplasia cutis congenita. Group 7 This is aplasia cutis congenita localized to the extremities without EB. At least two families have been reported in which multiple members have had extensive aplasia cutis congenita on the pretibial lower extremities and the dorsal aspects of the hands and the feet. Group 8 This is aplasia cutis congenita due to teratogens. A few cases of aplasia cutis congenita have been linked to intrauterine infection with herpes simplex virus or varicella zoster virus or to exposure to methimazole in the treatment of maternal thyrotoxicosis during pregnancy. Imperforate anus has been associated with methimazole or carbimazole exposure during gestation. Group 9 This is aplasia cutis congenita associated with malformation syndromes. Aplasia cutis congenita has been reported as a characteristic in many syndromes, including trisomy 13 (Patau syndrome) with large membranous scalp defects; 4p- (Wolf-Hirschhorn) syndrome with midline scalp defects; Setleis syndrome with bitemporal aplasia cutis congenita and abnormal eyelashes; Johanson-Blizzard syndrome with stellate scalp defects; focal dermal hypoplasia (Goltz syndrome); amniotic band disruption complex; oculocerebrocutaneous (Delleman) syndrome; scalp-ear-nipple syndrome (Finlay-Mark syndrome) ; Kabuki syndrome ; and 46XY gonadal dysgenesis. Reticulolinear aplasia cutis congenita on the face and neck is a distinctive cutaneous manifestation in several syndromes linked to band Xp22. There is no one cause for all cases of aplasia cutis congenita.The condition is thought to be multifactorial, which means that several factors likely interact to cause the condition.Factors that may contribute include genetic factors; teratogens (exposures during pregnancy that can harm a developing fetus) such as methimazole, carbimazole, misoprostol, and valproic acid; compromised vasculature to the skin; and trauma.Some cases may represent an incomplete or unusual form of a neural tube defect.Familial cases of aplasia cutis congenita have been reported.Cases that appear to be genetic may be inherited in an autosomal dominant or autosomal recessive manner.
