



Primary Hyperoxaluria
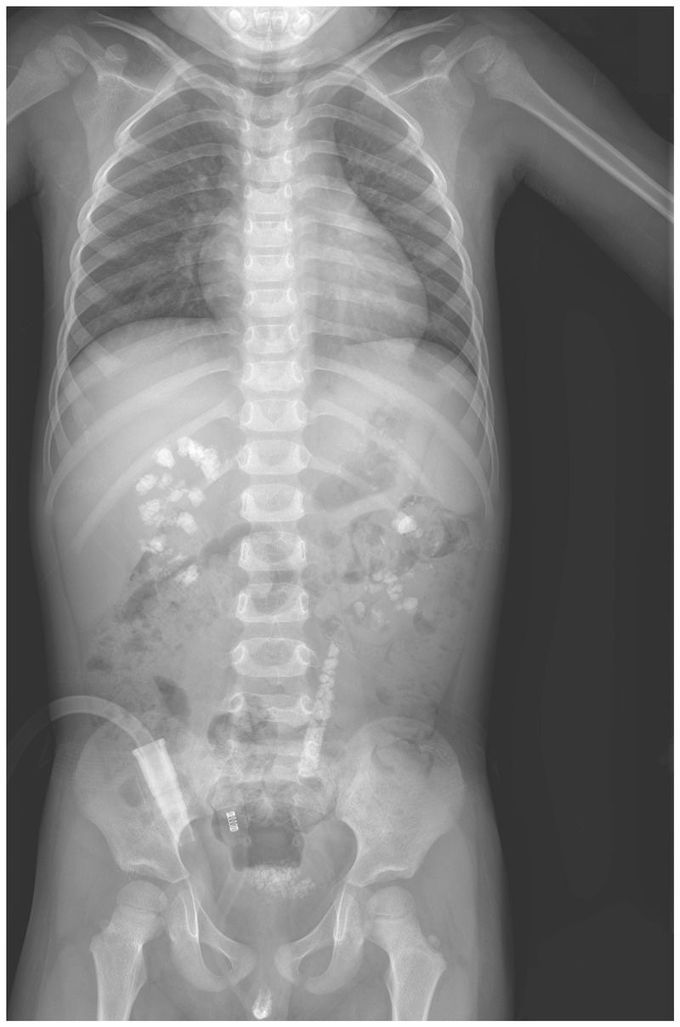
A 3-year-old boy presented to the pediatric urology clinic with a 6-month history of gross hematuria and intermittent abdominal pain. Urinalysis revealed red cells, white cells, and 3+ protein. The serum creatinine level was 0.9 mg per deciliter (80 μmol per liter; normal range in 3-year-old Chinese children, 0.3 to 0.8 mg per deciliter [20 to 70 μmol per liter]). The 24-hour urinary excretion of oxalate was elevated. A radiograph of the abdomen showed stones in the kidneys, bladder, and left ureter. Constituent analysis of the excreted stones indicated that the composition of the stones was more than 95% calcium oxalate monohydrate. On the basis of the clinical data, primary hyperoxaluria type 1 was suspected. Mutational analysis of AGXT, the gene encoding alanine–glyoxylate aminotransferase, confirmed the diagnosis in this patient. Primary hyperoxaluria type 1 is a rare autosomal recessive disorder of glyoxylate metabolism that leads to recurrent urolithiasis alone or in combination with nephrocalcinosis. Supportive measures were initiated, including high fluid intake and oral potassium citrate and vitamin B6. During 1 year of follow-up, the patient continued to have elevated levels of urinary oxalate and multiple kidney stones.
