



Primary Hyperoxaluria
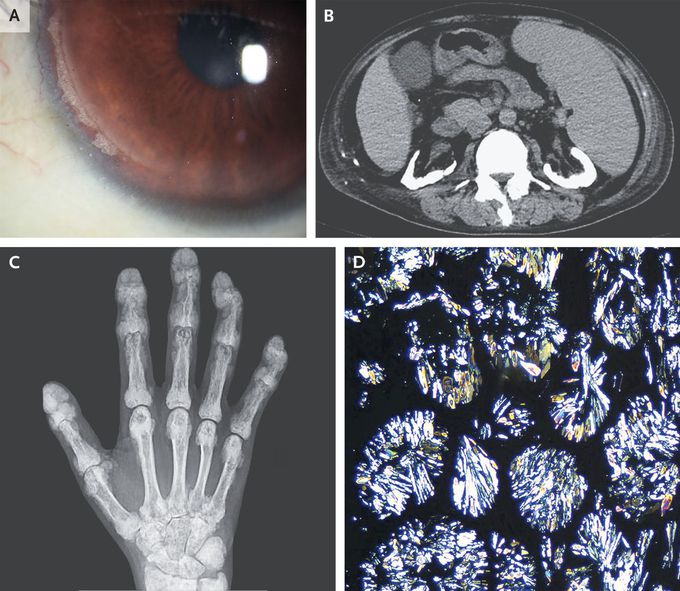
A 40-year-old man with end-stage kidney disease and calcium oxalate nephrolithiasis presented to the emergency department with a 7-year history of joint pain. He also reported blurry vision, painful skin lesions, and tooth loss, all of which had developed over the past several years. Physical examination was notable for the absence of incisors and the presence of synovitis of the elbows, knees, and ankles; deformities of the finger joints; and tender subcutaneous nodules on the back and the legs. Corneal calcium deposits were seen on slit-lamp examination (Panel A). Computed tomography of the abdomen showed nephrocalcinosis in both kidneys (Panel B), and radiography of the hands showed dense metaphyseal bands and calcium deposits in the soft tissues (Panel C). A biopsy specimen of a subcutaneous nodule showed calcium oxalate deposition with birefringence on polarized microscopy (Panel D). Two AGXT variants were observed on genetic testing, and a diagnosis of primary hyperoxaluria type 1 was made. Primary hyperoxaluria is a rare autosomal recessive disorder involving the overproduction of oxalate by the liver that may go undiagnosed for years. Early symptoms include nephrocalcinosis and nephrolithiasis. As kidney function worsens, systemic oxalate deposition may occur, including in the joints, bones, eyes, and skin. The patient had been undergoing hemodialysis, and his joint pain abated when the frequency of treatment was increased. He is undergoing evaluation for liver and kidney transplantation.
