



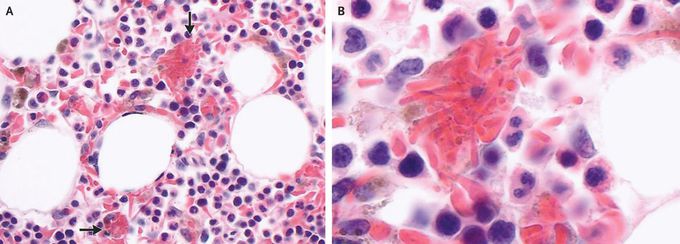
Hemophagocytosis of Sickle Cells
A 60-year-old man presented to the hematology clinic with fatigue and dyspnea on exertion. He had a history of sickle cell disease with hemoglobin genotype SS. Physical examination was notable for conjunctival pallor and mild scleral icterus. Laboratory studies showed a hemoglobin level of 5.9 g per deciliter (baseline range, 6 to 8; reference range, 13.5 to 16) with undetectable hemoglobin S. The ferritin level was elevated at 11,505 ng per milliliter (reference range, 22 to 277). The patient was admitted to the hospital, and evaluation for infectious causes of anemia, including testing for cytomegalovirus, Epstein–Barr virus, and parvovirus, was negative. Bone marrow biopsy revealed normocellular bone marrow with marked erythroid hyperplasia and hemosiderosis. Numerous hemophagocytic histiocytes containing sickle cells were seen on light microscopy (Panel A, arrows; Panel B shows a magnified view). The criteria for the diagnosis of hemophagocytic lymphohistiocytosis were not met. Hemophagocytosis in sickle cell disease is a rare but recognized entity that is associated with viral infections. Selective hemophagocytosis of sickle cells with peripheral disappearance of hemoglobin S as a cause of severe anemia is highly unusual. The patient received treatment with red-cell transfusions and iron chelation. He was discharged from the hospital and continues to receive maintenance red-cell transfusions every 3 weeks.
