



Leprechaunism / Donohue syndrome
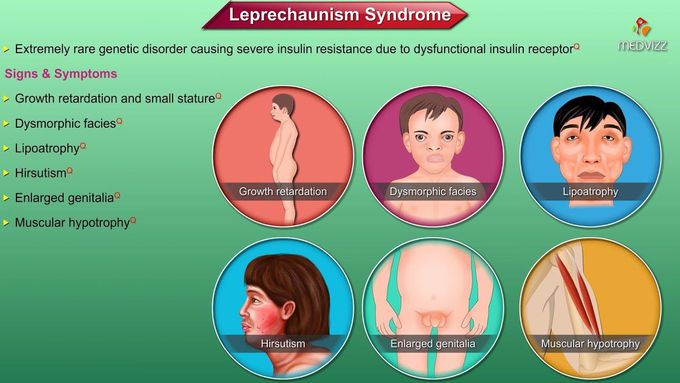
Leprechaunism / Donohue syndrome is an extremely rare and very severe genetic disorder in which the prime abnormality lies in defective formation of the insulin receptor. It is inherited in an autosomal recessive fashion. Babies born with this disease have craniofacial anomalies, low birth weight, skin abnormalities and genital enlargement. They may even lack muscle mass. Typical facial features include abnormally large, low-set and underdeveloped ears; flat bridge of the nose; large, thick lips; an abnormally large mouth (macrostomia); and widely spaced eyes (hyptertelorism). Affected infants may also have an abnormally small head (microcephaly). Leprechaunism is associated with abnormal darkening and thickening of patches of skin in certain areas of the body (acanthosis nigricans), unusual thickening of the skin (pachyderma), excessive hair growth (hirsutism), and malformation (dysplasia) of the nails. In addition, infants with leprechaunism exhibit absence of most of the body fat under the skin (subcutaneous adipose tissue). Leprechaunism is also characterized by abnormalities of the endocrine system (i.e., the system of glands that secrete hormones into the blood system). Such abnormalities include excessive secretion of the hormone insulin (hyperinsulinemia). Insulin regulates blood sugar (glucose) levels by promoting the movement of glucose into bodily cells. Infants with leprechaunism fail to use insulin effectively (insulin resistant). Due to the mutation in the insulin receptor gene, individuals with leprechaunism are unable to use insulin effectively. Insulin is a hormone produced by the pancreas that plays an important role in the absorption of sugar (glucose) into muscle cells. Glucose is the body’s main source of energy. Some symptoms associated with leprechaunism, including growth deficiencies and hyperglycemia, develop as a result of severe insulin resistance of affected individuals.
