



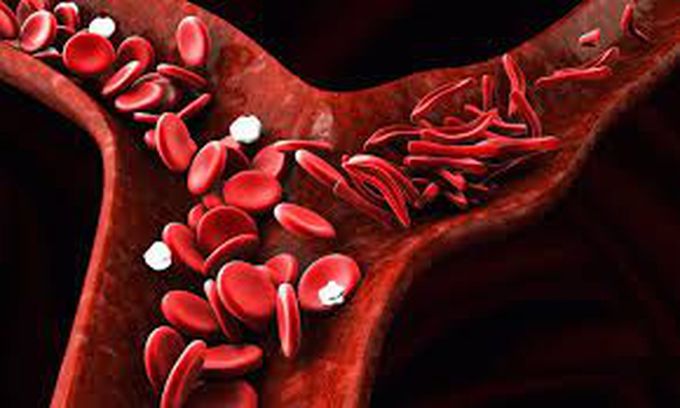
How to Manage Sickle Cell Crisis
Sickle cell disease is a group of genetic disorders that cause aberration in hemoglobin, resulting in decreased oxygen-carrying capacity. The consequent hypoxia may trigger a painful condition, commonly known as ‘sickle cell crisis’. Other complications may also include stroke, meningitis, and avascular necrosis etc. The most dreaded presentation is vaso-occlusion, which requires immediate management. Assessment of the patient and administration of oral / IV / intranasal analgesia is the first and foremost step in management, Vital signs should be observed and recoded carefully. Oxygen saturation should particularly be monitored vigilantly. Once the pain has subsided, the patient may be discharged. However, in cases where pain has not receded, the patient should be hospitalized. Stronger analgesics are recommended in such cases. Exchange transfusion may be indicated according to patient condition. Additional remedy may include ensuring sufficient hydration, use of hydroxyurea and anxiolytics. Once the symptoms have subsided, a consultation with hematologist is advised in order to identify the trigger factors and prevent future sickle cell crisis. Vaso-occlusive crisis should be treated immediately; delayed treatment may result in death. Source: Sickle Cell Crisis https://www.ncbi.nlm.nih.gov/books/NBK526064/ Image taken from https://www.drugtargetreview.com/news/33558/hri-depletion/
